計算機技術的高速發展,使得電腦及配套服務成為現代生活和工作中不可或缺的一部分。從購買到使用,再到維護與升級,用戶往往需要多環節的專業支持。為此,將電腦銷售、網絡安裝、軟硬件維修以及軟硬件研發整合為一體,可提供全面、高效的服務體驗。
電腦銷售是基礎環節。這不僅包括傳統臺式機、筆記本的銷售,還涉及定制化主機、服務器等設備。在銷售過程中,專業人員可根據用戶需求(如辦公、游戲、設計等)推薦合適的配置,并提供售后保障。結合市場趨勢,銷售方還可引入最新產品,如輕薄本、高性能工作站等,滿足多樣化需求。
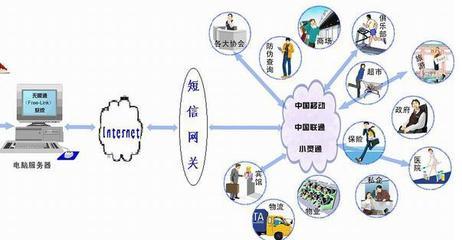
網絡安裝服務是連接用戶與數字世界的橋梁。無論是家庭網絡還是企業局域網,專業的安裝團隊可設計并部署穩定的網絡環境。這包括路由器配置、無線網絡覆蓋、網絡安全設置等。對于企業用戶,還可提供VPN、服務器搭建等高級服務,確保數據傳輸的安全與高效。
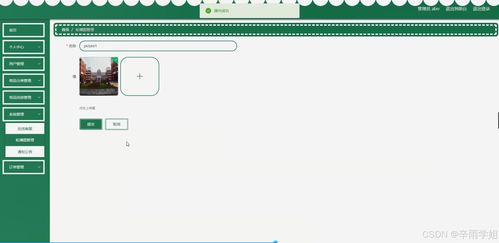
第三,軟硬件維修是保障設備長期運行的關鍵。硬件方面,常見問題如屏幕損壞、硬盤故障、電源問題等,可通過更換部件或維修解決;軟件方面,則涉及系統重裝、病毒清除、數據恢復等。通過快速響應和專業技術,維修服務可最大限度減少用戶停機時間,提升設備壽命。
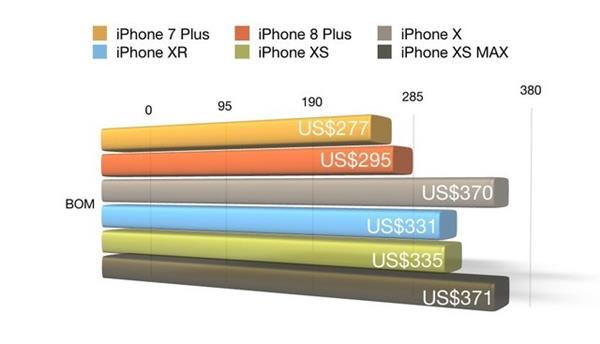
計算機軟硬件的研發是推動技術創新的動力。硬件研發可專注于優化性能、降低能耗,例如開發新型散熱系統或定制主板;軟件研發則包括操作系統優化、應用程序開發等,以滿足特定行業需求,如教育、醫療或金融領域。研發不僅提升服務深度,還可為銷售和維修提供技術支撐。
將電腦銷售、網絡安裝、軟硬件維修與研發整合,可形成閉環服務鏈,為用戶提供一站式解決方案。這不僅提高了效率,還增強了客戶粘性,是未來計算機服務行業的發展方向。